一种基于ONT测序的单细胞转录组的测序分析方法与流程
本发明涉及基因测序分析,特别涉及单细胞转录组的测序分析方法。
背景技术:
1、在生物医学研究和临床实践中,单细胞分析技术在揭示细胞异质性,解析复杂生物过程及疾病发病机制中正在发挥越来越关键的作用。传统的群体级分析方法掩盖了细胞间的细微差异,这些差异对于理解疾病发展、细胞决策过程以及对环境变化的响应至关重要。单细胞rna测序(scrna-seq)技术已经极大地推动了从单一细胞分辨成千上万个rna分子的检测和量化,为深入探索生物系统的内部机制提供了独特视角。
2、尽管scrna-seq技术取得了显著进展,但在捕获和分析全长rna分子方面的局限性,削弱了从单细胞获得的转录本完整信息,限制了对转录后修饰、可变剪接事件及基因表达调控机制的全面理解。
3、在此背景下,oxford nanopore technologies(ont)的单细胞全长测序技术应运而生,它通过直接测序单个细胞中的全长rna分子,提供了前所未有的转录本完整视角。这一技术不仅揭示了5’和3’末端、编码区、内含子和可变剪接事件,而且极大地拓宽了对单细胞基因表达复杂性的认识,为生物学研究和临床诊断提供了新的路径。
4、然而,ont单细胞全长测序技术在样本制备、数据处理和成本效益方面面临着挑战。这些挑战涉及保持rna分子的完整性、提升测序数据的质量和降低操作成本。因此,针对这些挑战开发新的优化方法,对扩大ont单细胞全长测序技术在科研和诊断中的应用具有至关重要的意义。
技术实现思路
1、本发明提供了一种基于ont测序的单细胞转录组的测序分析方法,本方法利用10×genomics单细胞捕获、标记技术和ont测序技术,能够更准确、更全面地分析细胞中的转录本和基因,深入挖掘生命过程中的转录信息,从而为生命科学研究、医学诊断和药物开发等领域提供更多有价值的数据和应用。本发明提供的单细胞转录组的测序分析方法具体通过以下技术实现。
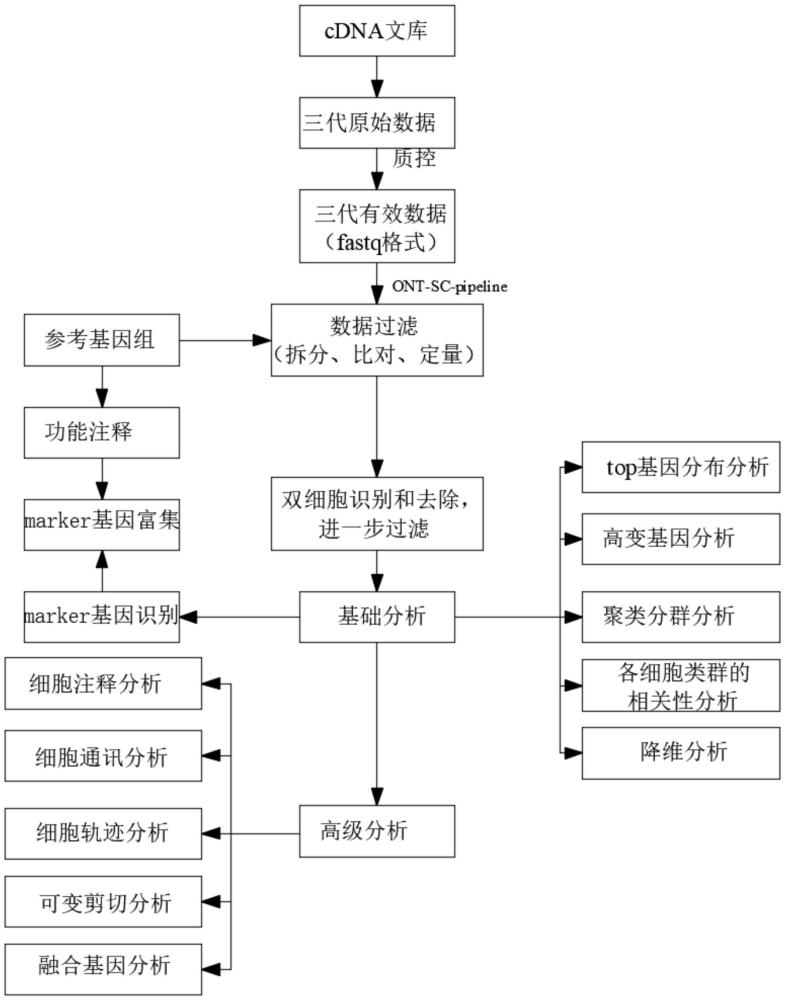
2、一种基于ont测序的单细胞转录组的测序分析方法,包括以下步骤:
3、制备、构建待测样品的cdna文库,测序,获得所述待测样品的三代原始数据;对所述三代原始数据进行质控,得到所述待测样品的三代有效数据;
4、对参考基因组的基因和转录本进行功能注释;
5、结合所述三代有效数据和所述参考基因组的基因数据,使用软件ont-sc-pipeline进行数据过滤、数据拆分、比对和定量,得到定量数据;
6、对所述定量数据进行双细胞识别和去除,并对数据进一步的过滤;
7、对过滤后的数据依次进行基础分析、marker基因富集分析和高级分析;所述基础分析包括top基因分布分析、高变基因分析、聚类分群分析、各细胞类群的相关性分析、降维分析和marker基因识别;所述高级分析包括细胞注释分析、细胞通讯分析、细胞轨迹分析、可变剪切分析和融合基因分析。
8、进一步地,所述质控处理的方法为:根据测序质量值进行筛选,剔除长度≤30bp和质量值≤5的序列。
9、进一步地,利用软件ont-sc-pipeline进行数据拆分、比对和定量的方法为:使用vsearch进行数据拆分,同时使用minimap2进行数据比对,提取拆分结果和比对结果二者信息合并得到所述定量数据。
10、进一步地,使用scdblfinder软件识别和去除双细胞和低活性细胞,并剔除核糖体rna比例较高、线粒体基因比例较高的细胞。
11、进一步地,使用seuratv5软件进行top基因分布分析;采用函数findvariablefeatures进行高变基因分析;使用seuratv5软件进行聚类分群分析、降维分析和marker基因识别。
12、更进一步地,所述降维分析具体采用seuratv5软件中的pca、t-sne、umap分析方法中的至少一种进行。
13、具体地,若待测样本为多样本,则使用harmony软件代替原本的pca分析方法,对过滤后的多样本单细胞数据进行校正和整合。
14、进一步地,采用超几何分布算法进行marker基因的富集分析。
15、进一步地,结合已知marker基因和scina软件进行细胞注释分析。
16、进一步地,采用cellphonedb v5进行细胞通讯分析。
17、进一步地,使用软件monocle3进行细胞轨迹分析。
18、进一步地,采用isoswitch软件进行可变剪切分析。
19、进一步地,采用软件jaffa进行融合基因分析。
20、进一步地,使用monocle3软件进行细胞轨迹分析。
21、进一步地,使用isoswitch软件进行可变剪切分析。
22、进一步地,使用jaffa软件进行融合基因分析。
23、进一步地,基于cellphonedb v5结果,利用ggplot2和igraph方法对单通路在各个细胞亚群之间的通讯结果进行可视化;将各亚基对配受体整体的贡献度和通路中单个配受体对的亚基进行可视化。
24、与现有技术相比,本发明的有益之处在于:
25、1、ont测序相比于二代测序手段,无需对片段进行打断,能够测到完整的转录本,能够对转录本进行定量。
26、2、相对于现有的pacbio三代测序而言更具有成本优势,在兼顾测序多种结果的同时具有极高的性价比。
技术特征:
1.一种基于ont测序的单细胞转录组的测序分析方法,其特征在于,包括以下步骤:
2.根据权利要求1所述的基于ont测序的单细胞转录组的测序分析方法,其特征在于,所述质控处理的方法为:根据测序质量值进行筛选,剔除长度≤30bp和质量值≤5的序列。
3.根据权利要求1所述的基于ont测序的单细胞转录组的测序分析方法,其特征在于,利用软件ont-sc-pipeline进行数据拆分、比对和定量的方法为:使用vsearch进行数据拆分,同时使用minimap2进行数据比对,提取拆分结果和比对结果二者信息合并得到所述定量数据。
4.根据权利要求1所述的基于ont测序的单细胞转录组的测序分析方法,其特征在于,使用scdblfinder软件识别和去除双细胞和低活性细胞,并剔除核糖体rna比例较高、线粒体基因比例较高的细胞。
5.根据权利要求1所述的基于ont测序的单细胞转录组的测序分析方法,其特征在于,使用seuratv5软件进行top基因分布分析;采用函数findvariablefeatures进行高变基因分析;使用seuratv5软件进行聚类分群分析、降维分析和marker基因识别。
6.根据权利要求5所述的基于ont测序的单细胞转录组的测序分析方法,其特征在于,所述降维分析具体采用seuratv5软件中的pca、t-sne、umap分析方法中的至少一种进行。
7.根据权利要求6所述的基于ont测序的单细胞转录组的测序分析方法,其特征在于,若待测样本为多样本,则使用harmony软件代替所述pca分析方法,对过滤后的多样本单细胞数据进行校正和整合。
8.根据权利要求1所述的基于ont测序的单细胞转录组的测序分析方法,其特征在于,采用超几何分布算法进行marker基因的富集分析。
9.根据权利要求1所述的基于ont测序的单细胞转录组的测序分析方法,其特征在于,结合已知marker基因和scina软件进行细胞注释分析;采用cellphonedb v5进行细胞通讯分析;使用软件monocle3进行细胞轨迹分析;采用isoswitch软件进行可变剪切分析;采用软件jaffa进行融合基因分析;使用monocle3软件进行细胞轨迹分析;使用isoswitch软件进行可变剪切分析;使用jaffa软件进行融合基因分析。
10.根据权利要求9所述的基于ont测序的单细胞转录组的测序分析方法,其特征在于,基于cellphonedb v5结果,利用ggplot2和igraph方法对单通路在各个细胞亚群之间的通讯结果进行可视化;将各亚基对配受体整体的贡献度和通路中单个配受体对的亚基进行可视化。
技术总结
本发明公开了一种基于ONT测序的单细胞转录组的测序分析方法,涉及基因测序分析技术领域。本方法包括以下步骤:制备、构建待测样品的cDNA文库,测序,获得三代原始数据;对三代原始数据进行质控,得到三代有效数据;对参考基因组进行功能注释;使用软件ONT‑SC‑pipeline进行数据拆分、比对和定量,得到定量数据;对定量数据依次进行基础分析、Marker基因富集分析和高级分析;基础分析包括top基因分布分析、高变基因分析、聚类分群分析、各细胞类群的相关性分析、降维分析和Marker基因识别;高级分析包括细胞注释分析、细胞通讯分析、细胞轨迹分析可变剪切分析和融合基因分析。
技术研发人员:冀金龙,田朝阳,宋驰,陈虎,杨路路,易欣欣
受保护的技术使用者:武汉贝纳科技有限公司
技术研发日:
技术公布日:2024/12/10
技术研发人员:冀金龙,田朝阳,宋驰,陈虎,杨路路,易欣欣
技术所有人:武汉贝纳科技有限公司
备 注:该技术已申请专利,仅供学习研究,如用于商业用途,请联系技术所有人。
声 明 :此信息收集于网络,如果你是此专利的发明人不想本网站收录此信息请联系我们,我们会在第一时间删除