一种基于密度曲线的DNA拷贝数检测方法及系统与流程
本发明涉及生物信息学,具体涉及一种基于密度曲线的dna拷贝数检测方法及系统。
背景技术:
1、近年来,拷贝数变异测序(cnv-seq),特别是单细胞cnv-seq(sccnv-seq)揭示了癌症演化及胚胎基因突变在全套基因组(染色体)和单个染色体及亚染色体水平(或dna节段)的突变图景。从测序数据中识别cnv一直是一个长期的挑战。目前的技术包括芯片杂交方法、中期染色体核型分析方法、和集团平行测序(特别是二代测序方法cnv-seq,也包括某些三代测序方法)等。
2、在群体细胞bulk cnv-seq(bcnv-seq)中,经常存在不明确的拷贝数值(非整数倍的dna节段,即这个dna节段的倍数介于相对应的单倍体dna节段的整数倍之间),这通常被视为cnv的细胞异质性。在bcnv-seq的情况下,这种情况可能代表群体细胞的异质性或嵌合体(chimeric,mosaic)如胚胎嵌合体。
3、对单细胞拷贝数测序(sccnv-seq)来说,无论这个细胞是整倍体、非整倍体,缺体、单体、三体或四体,理论上不存在相应单倍体dna节段的非整数倍的dna节段,而一定是单拷贝dna的整数倍,我们称整数型拷贝数(integer copy number)。
4、目前在诸多sccnv-seq报告中采用了对倍性数据进行四舍五入调整的策略来确定染色体或dna节段的单体、二体或三体。基本上是人为地省略小于0.5的数值的小数部分,并确定该数值以下的整数倍;当一个值的小数部分大于0.5时,确定该值的上一个整数倍,即是四舍五入的方法。我们发现,在sccnv-seq中的相对理想的场景下,舍入调整通常表现良好;但在许多其他实际临床案例中很难做出客观判断,即出现dna节段非整倍体的倍数的估计常常出现较大的人为误差(artificial error)。
5、所以,正确估计dna拷贝数是非常重要地,胚胎的客观准确的染色体倍数的判断决定临床措施的选择。
技术实现思路
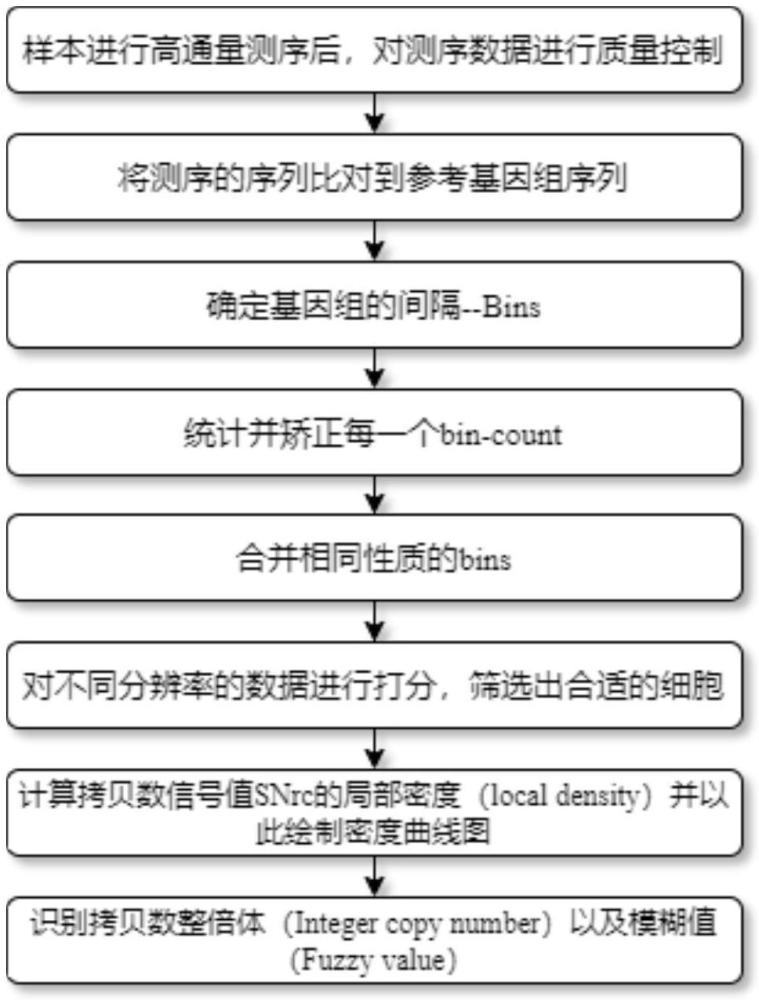
1、为了解决这个问题,本文提出的一种基于集团平行测序(massive parallel)数据的密度曲线估计客观拷贝数(即拷贝数的实际数量)的计算方法,充分考虑了实验误差和数据噪声的情况,引入模糊值这一概念(fuzzy value),保留了非整数型拷贝数的不确定性,用此方法进行基因拷贝数估计可以提供更准确、更客观的结果(工作流程参见图1)。
2、为实现上述目的,本发明采取的技术方案为:一种基于集团平行测序(massiveparallel)数据的密度曲线客观并准确判定拷贝数变异的计算方法和系统,包括如下步骤:
3、s1、将测序数据过滤,进行上游分析,输出原始数据;
4、s2、把相应生物的基因组划分为不重叠的窗口,所述窗口作为一个分割单位,称为一个bin区;
5、s3、统计每个bin区的reads读数,将得到原始覆盖数bincount;
6、s4、将bincount进行归一化处理,生成标准化的拷贝数nrc值(normalized readcount);
7、s5、通过算法工具,将具有近似nrc值(normalized read count)的连续bin区划分为同一个segment,并将拷贝数信号值snrc(segmented normalized read counts)作为能代表该segment区的所有nrc的集中趋势的量数;
8、s6、对不同预设分辨率的拷贝数信号值snrc数据进行打分,并根据经验设定的阈值(cut off)为标准,对符合标准的合格snrc数据进行下一步分析;
9、s7、计算拷贝数信号值snrc的局部密度(local density)并以此绘制密度曲线图;
10、s8、基于密度曲线图来找出主峰区(major peak zone)和缓冲区(buffer zone)分别作为整数型拷贝数(integer copy number)和模糊值(fuzzy value)
11、进一步地,对单细胞和群体细胞通过二代测序平台所产生的原始数据(raw data)进行质控和过滤后,输出可分析的数据(clean data)。
12、进一步地,根据相应生物的基因组序列进行映射,把基因组划分割为不重叠的窗口,所述窗口作为一个分割单位,称为一个bin区。
13、优选地,所述基因组为人类基因组,所述bin区的数量从1kbins到100k bins,对应的bin区长度约为3000kbp到30kbp。
14、进一步地,用python的varbin程序,根据hg19或其他版本的基因组序列的独特的可映射性(即测序得到的reads能唯一匹配的序列),人类基因组被分为不同大小的分割单位bins。其中varbin算法在仿真过程中所采用的模拟数据fastq文件的读长(reads)与后续测序所得到的读长(reads)应保持一致或近似一致。
15、优选地,可选择gingo、hmm copy等程序替代varbin程序。
16、进一步地,统计每个bin区的reads读数,将得到bincount进行归一化处理,生成标准化的拷贝数nrc值(normalized read count)。需要说明的是,考虑测序中的gc偏好的影响,即基因组上gc含量在50%左右的区域更容易被测到,产生的reads更多,这些区域的覆盖度更高,在高gc或者低gc区域,不容易被测到,产生较少的reads,这些区域的覆盖度更少,所以采用向每个bin的原始覆盖数(bincount)加上1,然后根据每个bins所在序列的gc含量使用lowess平滑方法对每个bin的原始覆盖数(bincount)进行归一化处理,生成标准化的拷贝数nrc(normalized read count)。
17、优选地,考虑到着丝粒和端粒附近存在重复序列会造成拷贝数虚高(显示高拷贝数,但是并不能反映真实的拷贝数)的情况,需要针对不同分辨率的bins下的数据,将着丝粒和端粒对应的序列所在bins的数据屏蔽,这种屏蔽通用于特定物种特定分辨率的任意数据。
18、进一步地,通过算法工具,将具有近似nrc值(normalized read count)的连续bin区划分为同一个segment,并能代表该segment区的所有nrc的集中趋势的量数称为拷贝数信号值snrc(segmented normalized read counts)。
19、优选地,算法工具包括但不限于kolmogorov-smirnov(ks)、循环二元分割算法circular binary segmentation(cbs)、高斯核平滑密度图(gaussian kernel smootheddensity plots)等程序,对“近似nrc值”的定义视具体程序工具而定。
20、优选地,能代表该segment区的所有nrc的集中趋势的量数包括但不限于算术平均数、几何平均数、中位数、众数。
21、进一步地,对不同预设分辨率的拷贝数信号值snrc数据进行打分,并根据经验设定的阈值(cut off)为标准,对符合标准的合格snrc数据进行下一步分析。
22、优选地,打分公式为:其中i表示第i个bin,n表示bins的总数,nrci和snrci分别代表第i个bin的nrc值和snrc值。我们选择nrcd小于0.25(作为缺省临界值:default threshold)的样本进行下一步分析。
23、较传统采用的cv值,提出的nrcd算法克服了传统采用的cv值会因为细胞发生cnv事件而变得虚高,从而更能有效衡量发生cnv时数据的波动性。
24、进一步地,计算拷贝数信号值snrc的局部密度并以此绘制密度曲线图。
25、优选地,根据snrc频率计算整个基因组的snrc密度分布,得到snrc频率的综合信息。公式如下所示:
26、
27、式中:fre(snrcbin_o)是snrc的频率,在集合nλ(bin_p)={bin_o|diff(bin_p,bin_o)<λ}中diff(bin_p,bin_o)表示第p个bin的数据与第o个bin的数据的差值,λ是差值的上限。需要说明的是,在计算局部密度时,所用的参数λ(滑动窗口)的宽度可以影响拷贝数信号值局部密度的峰图的平滑程度,参数λ(滑动窗口)越宽,局部密度的峰图越平滑,但所能识别出的波峰和波谷会相应减少。
28、进一步地,基于密度曲线图来找出主峰区(major peak zone)和缓冲区(bufferzone)分别作为整数型拷贝数(integer copy number)和模糊值(fuzzy value),具体指代见图2。
29、优选地,在密度图中的主峰区(majorpeak zone)为波峰(peak)和两侧波谷(vally)之间的区域,缓冲区(buffer zone)为相邻主峰区之间的区域
30、优选地,确定波峰和波谷的方法为,在密度图中对每一个snrc值进行判断是否为极值点,再按照筛选标准从中定义为波峰和波谷。
31、优选地,极值点的判断区间为所选snrc左右各延申0.01的范围。
32、优选地,选择合适的波峰和波谷要满足以下要求:1)波峰为极大值;2)波谷为极小值;3)波峰对应的密度值应大于1%;4)波峰的数量应小于或等于研究对象预期dna片段拷贝变异数的上限;5)波峰之间的距离应大致相等,优选地这个距离为1;6)波峰两侧的波谷之间的间隔(即主峰区的范围大小)应大于一个特定值,优选地这个特定值是0.5。
33、优选地,上述主峰的宽度即尺度(及主峰的数目)的确定原则以所研究样品的预估的全基因组范围内可能的染色体片段变异倍数的数目(一般为4-10以内)为限制范围。如,在人类数据中,存活胚胎和成人的染色体片段拷贝数目一般在1到4之间(除y染色体可能为0外)但是主要的是二倍体,相应的染色体片段的拷贝数是2;而未存活的(自然流产胚胎)或者植入前的胚胎虽然一般以二倍体为主,但是在个别情况下可能主要是三倍体或四倍体,变化范围更大;肿瘤的染色体片段拷贝数可能在0-10之间甚至变异范围更大,但是主要倍体数仍然是0-5倍体之间。因此,存活胚胎或成人,仅仅识别1到4个波峰,余此类推。
34、进一步的,所基于密度曲线的dna拷贝数检测系统包括:上游分析模块,用于对样本进行高通量测序后,对测序数据进行质量控制,对测序所得的分别进行过滤,去除掉接头序列和低质量碱基;序列比对模块,将序列比对到参考基因组,并对序列进行筛选以去除重复;滑动窗口模块,用于确定基因组的间隔—bins,以保证每一个bin具有相似的可映射性以及gc碱基含量;序列统计模块,与所述滑动窗口模块相连,对每个bin的序列进行统计计算并进行标准化;基因组分割模块,用以合并相同性质的bins;阈值确定模块,用于根据不同分辨率下的nrc和snrc数据确定样本数据的离散程度;密度计算模块,用于计算拷贝数信号值snrc的局部密度(local density)并以此绘制密度曲线图;峰值确定模块,用于寻找密度图中的波峰和波谷,并展示基因组上的拷贝数变异区域。
35、本发明还提供了计算机可读介质,记载有可以运行上述拷贝数变异的检测方法的程序。
36、与现有技术相比,本方案的有益效果:
37、相比传统cnv检测方法,本发明可与多种文库构建方法和通用的集团平行测序系统,包括二代测序、三代测序等,兼容,能够实现对细胞dna节段的拷贝数的准确识别;提出的nrcd算法较传统采用的cv值更能有效地衡量发生cnv时数据的波动性;可基于拷贝数的数据客观、准确、快速地对拷贝数进行dna节段的倍性进行客观识别;明确给出模糊值,便于判断单细胞测序sccnv-seq的样品是否存在非同一基因组细胞的dna污染,或者是群体细胞是否具有异致性或嵌合体的现象。
技术研发人员:潘星华,徐萌昌,林贯川
技术所有人:广州序科码生物技术有限责任公司
备 注:该技术已申请专利,仅供学习研究,如用于商业用途,请联系技术所有人。
声 明 :此信息收集于网络,如果你是此专利的发明人不想本网站收录此信息请联系我们,我们会在第一时间删除